Analisi delle immagini (bio) con Python Leggere e caricare immagini microscopiche utilizzando Matplotlib
Analisi immagini (bio) con Python lettura e caricamento immagini microscopiche con Matplotlib
Negli ultimi due decenni, il campo della microscopia ottica ha assistito a notevoli progressi grazie all’introduzione di tecniche innovative come la microscopia a scansione laser confocale (CLSM), la microscopia a scansione laser a fotoni multipli (MPLSM), la microscopia a fluorescenza a luce di foglio (LSFM) e la microscopia ad alta risoluzione [1]. Queste tecniche hanno notevolmente migliorato la capacità di acquisire immagini con una maggiore penetrazione in profondità, vitalità e risoluzione. Tuttavia, insieme a questi notevoli progressi, l’emergere di queste tecniche sofisticate ha presentato notevoli sfide nella gestione delle enormi quantità di dati generati. La visualizzazione, l’analisi e la diffusione efficace di questi complessi dataset sono diventate un aspetto cruciale per sfruttare appieno il potenziale di queste tecniche di microscopia all’avanguardia. In questo tutorial, esploreremo come leggere e visualizzare immagini complesse di microscopio ottico 2D, 3D, 4D e 5D utilizzando la potente libreria Python Matplotlib.
Indice
- 1.1 Immagini e Pixel
- 1.2 Canali
- 1.3 Immagini 4D (x, y, canali e fette Z)
- 1.4 Proiezioni
- 1.5 Immagini 5D (x, y, canali, fette Z e tempo)
- 1.6 Conclusioni
- 1.7 Riferimenti
# Importa le librerieimport numpy as npimport matplotlib.pyplot as pltimport skimage as skfrom matplotlib_scalebar.scalebar import ScaleBarImmagini e Pixel
Le immagini microscopiche vengono registrate digitalmente e, per realizzare ciò, il flusso di fotoni che forma l’immagine finale deve essere diviso in piccole sotto-unità geometriche, gli elementi di immagine (pixel) [2]. Per quanto riguarda il computer, ogni pixel è solo un numero (Fig. 1), e quando i dati dell’immagine vengono visualizzati, i valori dei pixel vengono generalmente convertiti in quadrati; i quadrati non sono altro che una visualizzazione utile che ci consente di ottenere un’impressione rapida del contenuto dell’immagine [3].
![Figura 1. Immagine microscopica della localizzazione subcellulare della proteina eisosomale PilA-GFP [5], [6] nella conidiospora del fungo *Aspergillus nidulans* con un'area ingrandita (insetto rosso) che mostra i valori dei pixel. L'immagine è stata acquisita utilizzando un microscopio a multiphoton Leica TCS SP8 MP (figura fornita dall'autore).](https://miro.medium.com/v2/format:webp/1*L4Luyzm04ji5moFMT36iEw.png)
Nella microscopia e specificamente nella fluorescenza, le immagini microscopiche di solito vengono acquisite senza informazioni sul colore (utilizzando telecamere monocromatiche o fotomoltiplicatori) e vengono (di solito) visualizzate utilizzando una LUT (look-up table) a falso colore monocromatica. Le tabelle di ricerca (LUT; talvolta chiamate anche mappe a colori) determinano come i valori di grigio vengono tradotti in un valore di colore. Per visualizzare le immagini microscopiche, spesso utilizziamo tonalità di grigio (LUT grigia), oppure nel caso in cui vogliamo aumentare la sensibilità visiva del lettore alle caratteristiche deboli, invertiamo l’immagine, mappando i valori scuri su quelli chiari e viceversa [4].
Tutte le immagini microscopiche pubblicate dovrebbero contenere una barra di scala come riferimento visivo per la dimensione delle strutture o delle caratteristiche osservate nell’immagine.
- Hanno davvero bisogno di tutti quei livelli i grandi modelli di linguaggio? Questa ricerca sull’IA svela l’efficienza dei modelli La ricerca dei componenti essenziali nei grandi modelli di linguaggio
- Come perfezionare localmente un modello LLM per un compito di domanda e risposta (QA)
- GPT4Readability – Non scrivere mai più un README
# Leggi l'immagine utilizzando scikit-imageimg = sk.io.imread("Single_channel_eisosome.tif")# Stampa le dimensioni dell'immagineprint(f"Le dimensioni delle immagini sono: {img.shape[0]} x {img.shape[1]} pixel")# Crea una figura con due sottofigurefig, axs = plt.subplots(1, 2, figsize=(6, 4))# Traccia l'immagine in scala di grigi nella prima sottofiguraaxs[0].imshow(img,cmap="gray")axs[0].axis('off')# Aggiungi una barra di scala alla sottofigurascalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right')axs[0].add_artist(scalebar)axs[0].set_title("Immagine in scala di grigi")# Traccia l'immagine in scala di grigi invertita nella seconda sottofiguraaxs[1].imshow(img,cmap="gray_r")axs[1].axis('off')# Aggiungi una barra di scala alla sottofigurascalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right')axs[1].add_artist(scalebar)axs[1].set_title("Immagine invertita")# Regola lo spaziamento tra le sottofigureplt.tight_layout()Le dimensioni delle immagini sono: 589 x 589 pixel
Canali
Nella microscopia, i diversi canali rappresentano intervalli spettrali distinti utilizzati per acquisire e visualizzare componenti o strutture specifiche nel campione. Ogni canale corrisponde a un intervallo di lunghezza d’onda specifico o a uno spettro di emissione, consentendo ai ricercatori di visualizzare contemporaneamente diverse molecole, strutture o eventi all’interno del campione. Per ottimizzare il riconoscimento visivo, si consiglia l’uso di canali separati in bianco e nero per la maggior parte delle applicazioni di imaging fluorescente. È possibile creare un’immagine colorata finale fusa per mostrare l’overlay, garantendo l’accessibilità per i lettori affetti da forme comuni di daltonismo. Nel codice qui sotto, visualizziamo la localizzazione subcellulare di PilA-GFP (utilizzando una tabella di ricerca in scala di grigi) e Histone H1-mRFP (utilizzando una tabella di ricerca in scala di grigi), così come il loro overlay (PilA mostrato in magenta e Histone H1 in verde) [7] nella conidiospora di A. nidulans.
# Leggi l'immagine usando scikit-imageimg = sk.io.imread("3_channel_eisosome.tif")# Stampa le dimensioni dell'immagineprint(f"Le dimensioni delle immagini sono {img.shape[0]} x {img.shape[1]} pixel con {img.shape[2]} canali ")# Separa i canalih1_histone = img[:,:,0]pila = img[:,:,1]brightfield = img[:,:,2]# Crea una figura con quattro sottoplotfig, axs = plt.subplots(1, 4, figsize=(12, 4))# Plotta l'istogramma di H1 nel primo sottoplotaxs[0].imshow(h1_histone,cmap="gray")axs[0].axis('off')axs[0].set_title("H1 istone (Immagine in scala di grigi)")# Plotta il PilA nel secondo sottoplotaxs[1].imshow(pila,cmap="gray")axs[1].axis('off')axs[1].set_title("Proteina PilA (Immagine in scala di grigi)")# Plotta l'immagine brightfield nel terzo sottoplot con la barra di scala axs[2].imshow(brightfield,cmap="gray")axs[2].axis('off')scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right')axs[2].add_artist(scalebar)axs[2].set_title("Brightfield")# Converte le slice 2D in un array 3Dh1_histone_3d = np.atleast_3d(img[:,:,0]) #converte la slice 2D in un array 3Dpila_3d = np.atleast_3d(img[:,:,1]) #converte la slice 2D in un array 3D# Definisci il colorecolor_h1 = np.asarray([1, 0, 1]).reshape((1, 1, -1)) #crea un array 1x1x3 - colore magentacolor_pila = np.asarray([0, 1, 0]).reshape((1, 1, -1)) #crea un array 1x1x3 - colore verde# Moltiplica ogni pixel dell'immagine per il colore corrispondenteh1_magenta = h1_histone_3d * color_h1pila_green = pila_3d * color_pila# Fonde i canali coloratimerged = np.clip(h1_magenta + pila_green,0,255)# Plotta i canali fusi nel quarto sottoplotaxs[3].imshow(merged)axs[3].axis('off')axs[3].set_title("Canali fusi")# Aderisce lo spazio tra i sottoplotplt.tight_layout()Le dimensioni delle immagini sono 589 x 589 pixel con 3 canali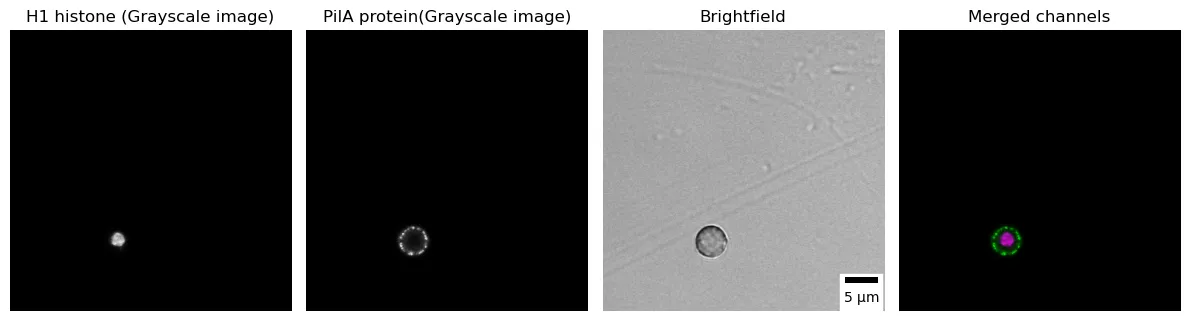
Immagini 4D (x,y, canali e Z-slices)
Molto spesso nella microscopia confocale, acquisiamo “slices” di immagini a diversi piani focali lungo l’asse Z (uno stack Z). Queste slices vengono quindi ricostruite in un’immagine 3D impilandole insieme. Gli stack Z forniscono preziose informazioni sulla distribuzione spaziale, la morfologia e le relazioni delle strutture all’interno del campione in tre dimensioni. Nel codice qui sotto, dimostriamo la visualizzazione della localizzazione subcellulare di PilA-GFP e Histone H1-mRFP nella conidiospora di A. nidulans acquisendo quattro “slices” lungo l’asse Z (Nella pratica, l’immagine sottostante rappresenta un iperstack, una composizione 4D composta da x, y, z e canali). Questo approccio ci consente di studiare la distribuzione e le relazioni di queste proteine in tre dimensioni.
# Leggere l'immagine utilizzando scikit-imageimg = sk.io.imread("3_channel_zstack_eisosome.tif")# Stampare le dimensioni dell'immagineprint(f"Le dimensioni dell'immagine sono {img.shape[1]} x {img.shape[2]} pixel con {img.shape[3]} canali e {img.shape[0]} strati")# Separare i canalih1_histone = img[:,:,:,0]pila = img[:,:,:,1]brightfield = img[:,:,:,2]# Creare una figura con sottoplotfig, axs = plt.subplots(3, 4, figsize=(12, 7))# Tracciare le singole immagini del canale H1 histonefor i in range(img.shape[0]): axs[0][i].imshow(h1_histone[i,:,:],cmap="gray") axs[0][i].axis('off') axs[0][i].set_title(f"(Z = {i+1}) H1 histone") scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right') axs[0, -1].add_artist(scalebar) # Tracciare le singole immagini del canale PilA protein for i in range(img.shape[0]): axs[1][i].imshow(pila[i,:,:],cmap="gray") axs[1][i].axis('off') axs[1][i].set_title(f"(Z = {i+1}) PilA protein") scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right') axs[1, -1].add_artist(scalebar)# Creare immagini unite dei canali H1 histone e PilA proteinfor i in range(img.shape[0]): h1_histone_3d = np.atleast_3d(h1_histone[i,:,:]) #converte la fetta 2D in un array 3D pila_3d = np.atleast_3d(pila[i,:,:]) #converte la fetta 2D in un array 3D # Definire il colore color_h1 = np.asarray([1, 0, 1]).reshape((1, 1, -1)) #crea un array 1x1x3 - colore magenta color_pila = np.asarray([0, 1, 0]).reshape((1, 1, -1)) #crea un array 1x1x3 - colore verde h1_magenta = h1_histone_3d * color_h1 pila_green = pila_3d * color_pila merged = np.clip(h1_magenta + pila_green,0,255) axs[2][i].imshow(merged) axs[2][i].axis('off') axs[2][i].set_title(rf"(Z = {i+1}) H1 e PilA") scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right') axs[2, -1].add_artist(scalebar)# Regolare lo spaziamento tra i sottoplotplt.tight_layout()Le dimensioni dell'immagine sono 589 x 589 pixel con 3 canali e 4 strati
Proiezioni
Un modo per visualizzare uno stack Z è quello di esaminare ogni singolo strato individualmente, come dimostrato nel codice sopra. Tuttavia, questo approccio diventa scomodo quando si lavora con più strati. Un metodo efficiente per riassumere le informazioni in uno stack Z è calcolare una proiezione Z [3]. Effettuando una proiezione Z, possiamo generare vari tipi di proiezioni in base alla domanda specifica a cui vogliamo rispondere. Ad esempio, possiamo ottenere una proiezione massima, minima, media o di deviazione standard. In una proiezione massima, viene creata un’immagine 2D per rappresentare l’intensità massima, catturando la caratteristica più luminosa, lungo un asse specifico, tipicamente l’asse Z. Allo stesso modo, nelle proiezioni media, minima e di deviazione standard, generiamo immagini 2D che rappresentano rispettivamente la media, il minimo o la dispersione delle intensità lungo un asse specifico.
# Leggere l'immagine utilizzando scikit-imageimg = sk.io.imread("3_channel_zstack_eisosome.tif")# Separare i canalih1_histone = img[:,:,:,0]pila = img[:,:,:,1]brightfield = img[:,:,:,2]# Creare una figura con sottoplotfig, axs = plt.subplots(1, 4, figsize=(12, 7))# Lista contenente i nomi delle proiezioni da visualizzareprojections = ['Proiezione minima', 'Proiezione media', 'Proiezione massima', 'Proiezione deviazione standard']# Lista contenente i nomi delle proiezioni da calcolarenp_projection = [np.min, np.mean, np.max, np.std]# Calcolare le proiezioni; mantenere lo stesso dtype nell'outputfor i in range(len(projections)): h1_histone_3d = np.atleast_3d(np_projection[i](h1_histone, axis=0).astype(h1_histone.dtype)) #converte la fetta 2D in un array 3D pila_3d = np.atleast_3d(np_projection[i](pila, axis=0).astype(pila.dtype)) #converte la fetta 2D in un array 3D color_h1 = np.asarray([1, 0, 1]).reshape((1, 1, -1)) #crea un array 1x1x3 - colore magenta color_pila = np.asarray([0, 1, 0]).reshape((1, 1, -1)) #crea un array 1x1x3 - colore verde h1_magenta = h1_histone_3d * color_h1 pila_green = pila_3d * color_pila merged = np.clip(h1_magenta + pila_green,0,255) axs[i].imshow(merged) scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right') axs[i].add_artist(scalebar) axs[i].axis('off') axs[i].set_title(projections[i])
Immagini 5D (x, y, canali, fette Z e tempo)
L’imaging confocale 5D è uno strumento prezioso in biologia, che offre notevoli approfondimenti sui processi dinamici a livello cellulare e subcellulare. Queste immagini comprendono fette x, y, z e canali insieme al tempo, consentendo la visualizzazione e l’analisi dei cambiamenti temporali. Nel codice seguente, dimostriamo l’importanza dell’imaging confocale 5D presentando proiezioni massime di PilA-GFP e Histone H1-mRFP (immagine fusa) ad ogni istante di tempo, con ogni istante di tempo che rappresenta circa 20 minuti.
# Leggi l'insieme di immagini 5D utilizzando scikit-imageimg = sk.io.imread("3_channel_zstack_time_eisosome.tif")print(f"L'insieme di dati ha {img.shape[2]} x {img.shape[3]} pixel con {img.shape[4]} canali, {img.shape[1]} fette e {img.shape[0]} istanti di tempo")# Separa i canali h1_histone = img[:,:,:,:,0]pila = img[:,:,:,:,1]brightfield = img[:,:,:,:,2]# Crea una figura con sottoplotfig, axs = plt.subplots(6, 7, figsize=(19,14))# Calcola le proiezioni (mantenendo lo stesso dtype nell'output) e crea immagini fuse per ogni istante di tempotime =0for i in range (img.shape[0]): # Calcola la proiezione massima per il canale H1 histone h1_histone_3d = np.atleast_3d(np.max(h1_histone[i,:,:,:], axis=0).astype(h1_histone.dtype)) # Calcola la proiezione massima per il canale PilA pila_3d = np.atleast_3d(np.max(pila[i,:,:,:], axis=0).astype(pila.dtype)) # Calcola la proiezione minima per il canale brightfield brightfield_3d = np.atleast_3d(np.min(brightfield[i,:,:,:], axis=0).astype(brightfield.dtype)) # Definisci i colori per ogni canale color_h1 = np.asarray([1, 0, 1]).reshape((1, 1, -1)) #crea un array 1x1x3 - colore magenta color_pila = np.asarray([0, 1, 0]).reshape((1, 1, -1)) #crea un array 1x1x3 - colore verde color_brightfield = np.asarray([0.5, 0.5, 0.5]).reshape((1, 1, -1)) #crea un array 1x1x3 - colore grigio # Crea l'immagine fusa combinando i canali h1_magenta = h1_histone_3d * color_h1 pila_verde = pila_3d * color_pila brightfield_grigio = brightfield_3d* color_brightfield # fused = np.clip(h1_magenta + pila_green+brightfield_gray,0,255) fused = np.clip(h1_magenta + pila_verde,0,255)# Mostra l'immagine fusa nel sottoplot corrispondente axs[i // 7, i % 7].imshow(fused) axs[i // 7, i % 7].axis('off') axs[i // 7, i % 7].set_title(f"{time} min") scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right') axs[5, 1].add_artist(scalebar) time +=20plt.tight_layout()plt.show()L'insieme di dati ha 589 x 589 pixel con 3 canali, 4 fette e 37 istanti di tempo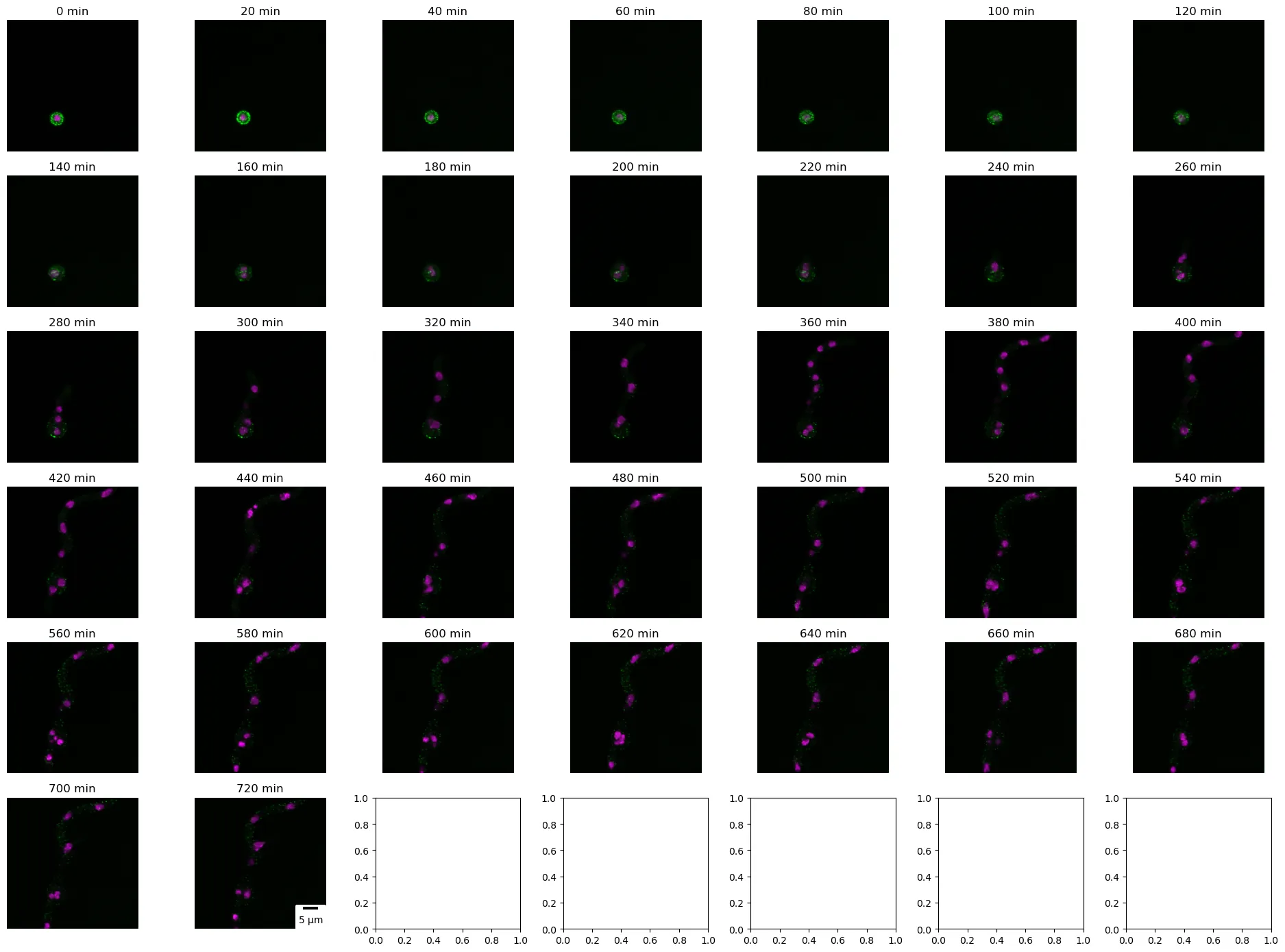
Visualizzazione (di seguito) di una fetta di brightfield del fungo A. nidulans a diversi istanti di tempo.
# Leggi l'insieme di immagini 5D utilizzando scikit-imageimg = sk.io.imread("3_channel_zstack_time_eisosome.tif")# Separa i canali h1_histone = img[:,:,:,:,0]pila = img[:,:,:,:,1]brightfield = img[:,:,:,:,2]# Crea una figura con sottoplotfig, axs = plt.subplots(6, 7, figsize=(19,14))# Calcola le proiezioni (mantenendo lo stesso dtype nell'output) e crea immagini fuse per ogni istante di tempotime =0for i in range (img.shape[0]):# Seleziona una fetta del canale Brightfield brightfield_2_slice = brightfield[i,2,:,:]# Mostra la seconda fetta nel sottoplot corrispondente axs[i // 7, i % 7].imshow(brightfield_2_slice, cmap="gray") axs[i // 7, i % 7].axis('off') axs[i // 7, i % 7].set_title(f"{time} min") scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right') axs[5, 1].add_artist(scalebar) time +=20plt.tight_layout()plt.show()

Conclusioni
In questo tutorial, approfondiremo il processo di lettura e visualizzazione di immagini intricate al microscopio ottico con varie dimensioni, tra cui 2D, 3D, 4D e 5D. Anche se matplotlib è una potente libreria per la visualizzazione dei dati, potrebbe non essere la scelta più adatta per la visualizzazione di immagini 3D, 4D e 5D, come le immagini microscopiche. Mentre matplotlib può gestire efficacemente le immagini 2D, la visualizzazione di immagini di dimensioni superiori può essere sfidante e limitata. Tuttavia, esistono soluzioni:
- Microfilm [9], [10], che semplifica la rappresentazione di immagini multicanale
- e Napari [8], una popolare libreria Python appositamente progettata per la visualizzazione e l’esplorazione di dati scientifici multidimensionali, comprese le immagini microscopiche. Napari fornisce un’interfaccia interattiva e versatile che va oltre le capacità di matplotlib.
Nel nostro prossimo tutorial, approfondiremo Napari ed esploreremo in dettaglio le sue caratteristiche per comprendere meglio le sue capacità di visualizzazione di immagini di dimensioni superiori.
Ho preparato un Jupyter Notebook da accompagnare a questo post del blog, che può essere visualizzato sul mio GitHub.

Riferimenti:
[1] C. T. Rueden and K. W. Eliceiri, “Visualization approaches for multidimensional biological image data,” BioTechniques, vol. 43, no. 1S, pp. S31-S36, lug. 2007, doi: 10.2144/000112511.
[2] J. B. Pawley, “Points, Pixels, and Gray Levels: Digitizing Image Data,” in Handbook Of Biological Confocal Microscopy, J. B. Pawley, Ed., Boston, MA: Springer US, 2006, pp. 59-79. doi: 10.1007/978-0-387-45524-2_4.
[3] P. Bankhead, “Introduction to Bioimage Analysis – Introduction to Bioimage Analysis.” https://bioimagebook.github.io/index.html (accessed 29 giu. 2023).
[4] J. Johnson, “Not seeing is not believing: improving the visibility of your fluorescence images,” Mol. Biol. Cell, vol. 23, no. 5, pp. 754-757, mar. 2012, doi: 10.1091/mbc.e11-09-0824.
[5] I. Vangelatos, K. Roumelioti, C. Gournas, T. Suarez, C. Scazzocchio, and V. Sophianopoulou, “Eisosome Organization in the Filamentous AscomyceteAspergillus nidulans,” Eukaryot. Cell, vol. 9, no. 10, pp. 1441-1454, ott. 2010, doi: 10.1128/EC.00087-10.
[6] A. Athanasopoulos, C. Gournas, S. Amillis, and V. Sophianopoulou, “Characterization of AnNce102 and its role in eisosome stability and sphingolipid biosynthesis,” Sci. Rep., vol. 5, no. 1, dic. 2015, doi: 10.1038/srep15200.
[7] A. P. Mela e M. Momany, “Diffusione internucleare dell’istone H1 all’interno dei compartimenti cellulari di Aspergillus nidulans,” PloS One, vol. 13, n. 8, p. e0201828, 2018, doi: 10.1371/journal.pone.0201828.
[8] “napari: un visualizzatore veloce e interattivo per immagini multidimensionali in Python – napari.” https://napari.org/stable/ (accesso il 04 luglio 2023).
[9] G. Witz, “microfilm.” 27 giugno 2023. Consultato il 14 luglio 2023. [Online]. Disponibile: https://github.com/guiwitz/microfilm
[10] “Bio-image Analysis Notebooks – Bio-image Analysis Notebooks.” https://haesleinhuepf.github.io/BioImageAnalysisNotebooks/intro.html (accesso il 14 luglio 2023).






